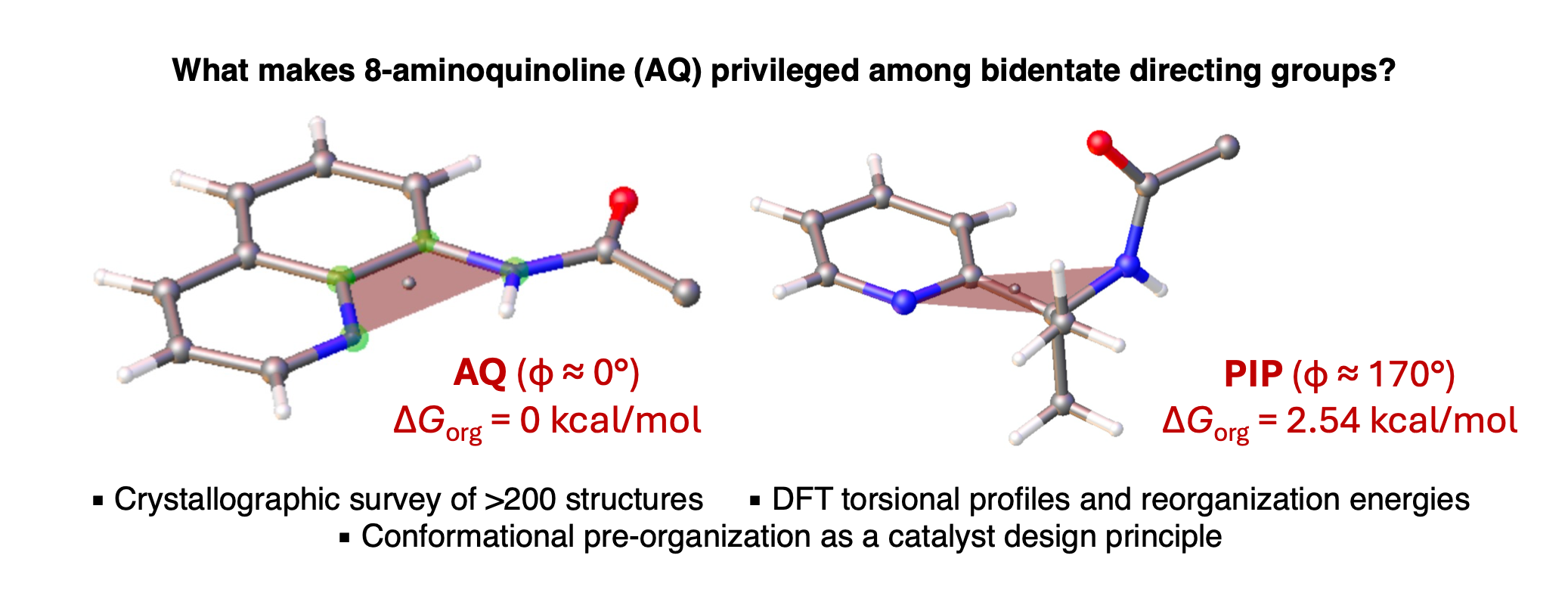
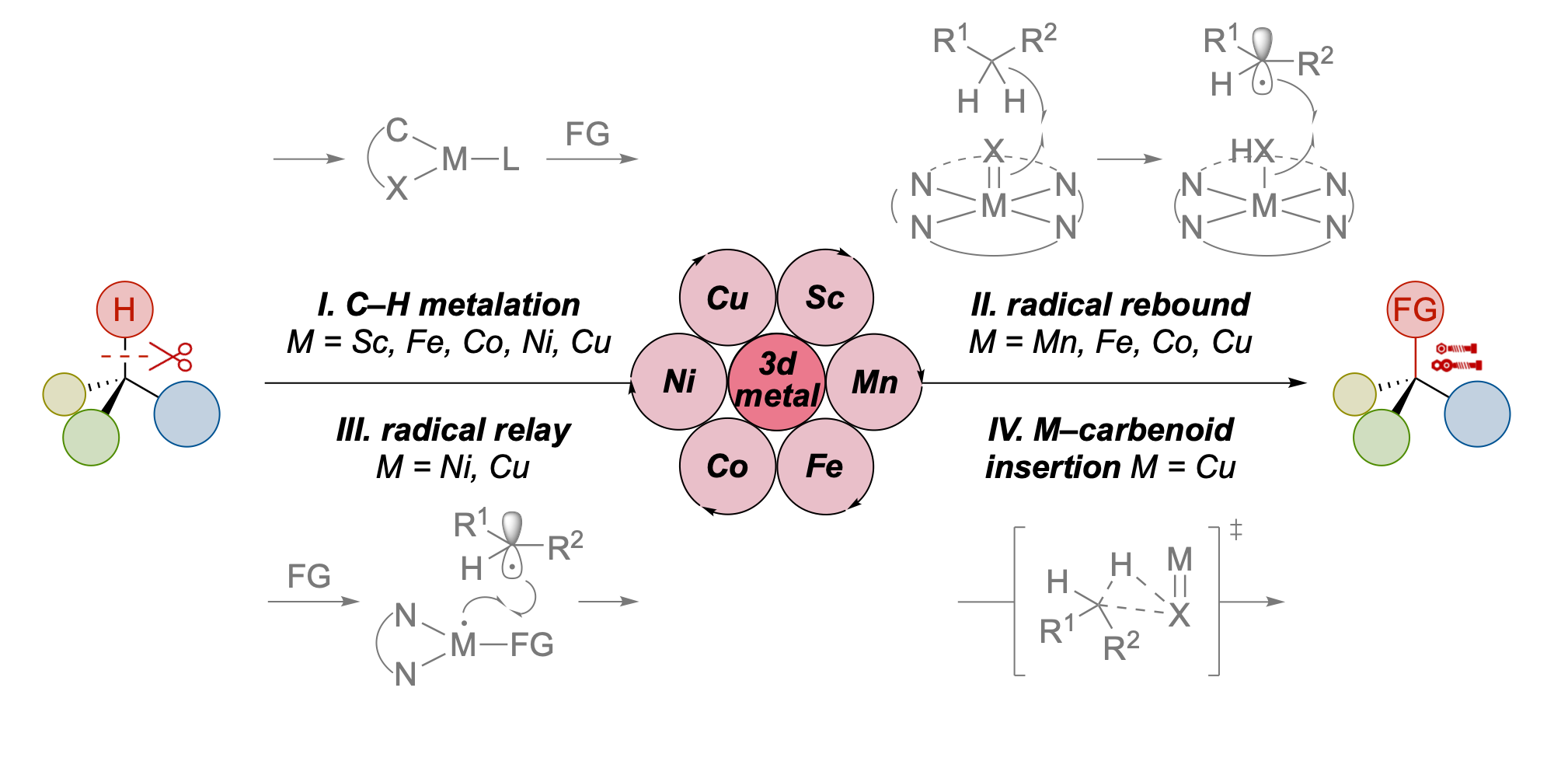
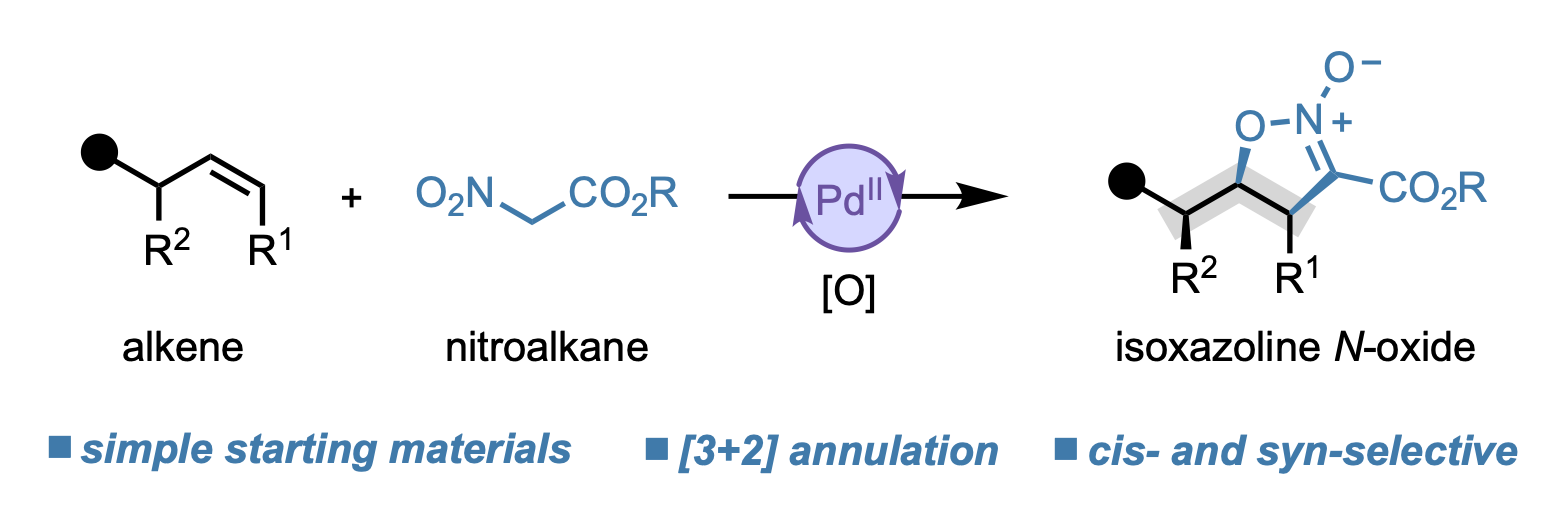
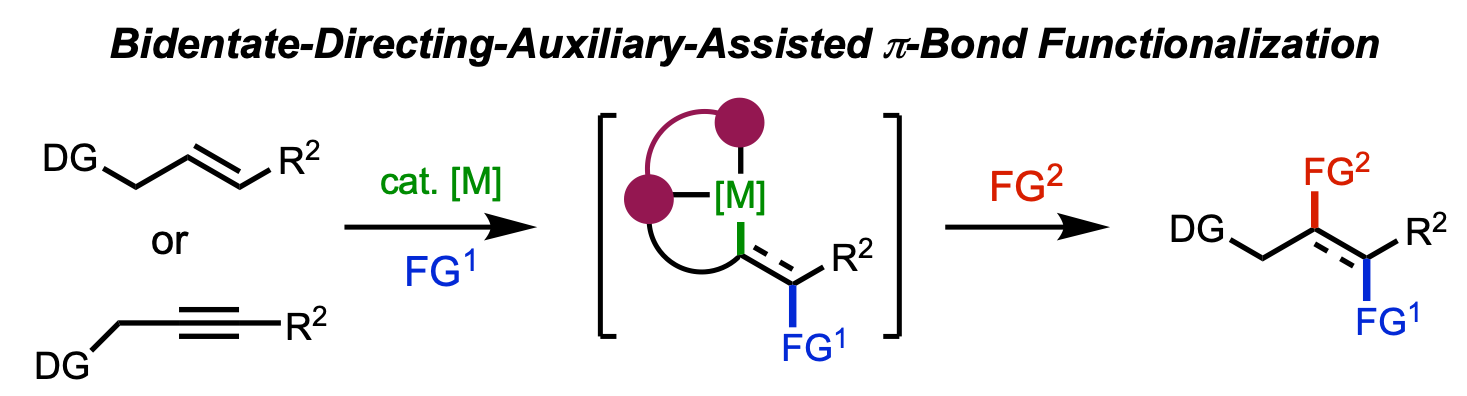
We are pleased to announce that the 2026 edition of our annual Organometallics Bootcamp begins next week. The short course is targeted to senior undergraduates and incoming graduate students, and we also welcome participation from chemists in industry, high school students, or anyone looking to grow their knowledge of organometallic chemistry. The course is highly flexible and accommodates in-person or virtual participation, synchronously or asynchronously.
The bootcamp runs Monday–Friday, August 10–21, with most classes held at 8:00 AM PT. Classes are held in hybrid format — in person at the Scripps Library Conference Room (and BCC 4th floor conference room on select days) and via Zoom. Certificates of completion are available upon request.
Click the link here to register: https://docs.google.com/forms/d/e/1FAIpQLSeGGFZkLqWzH_P2a0tFdv95NQX8aYtA7P8BFismRwo_vkZkxA/viewform